Log_Normal_TimeCourse.R is a tool developed in the R environment for statistical computing for
timepoint selection strategy for in vivo protein turnover from LC-MS data of heavy water metabolic labeling and LC-MS.
Longer labeling
duration and dense timepoint sampling (TPS) of tissues provide accurate proteome dynamics estimations.
However, the experiments are expensive. They require animal housing and care, as well as the labeling
with stable isotopes. It is, therefore, necessary to optimize the time points chosen for label sampling.
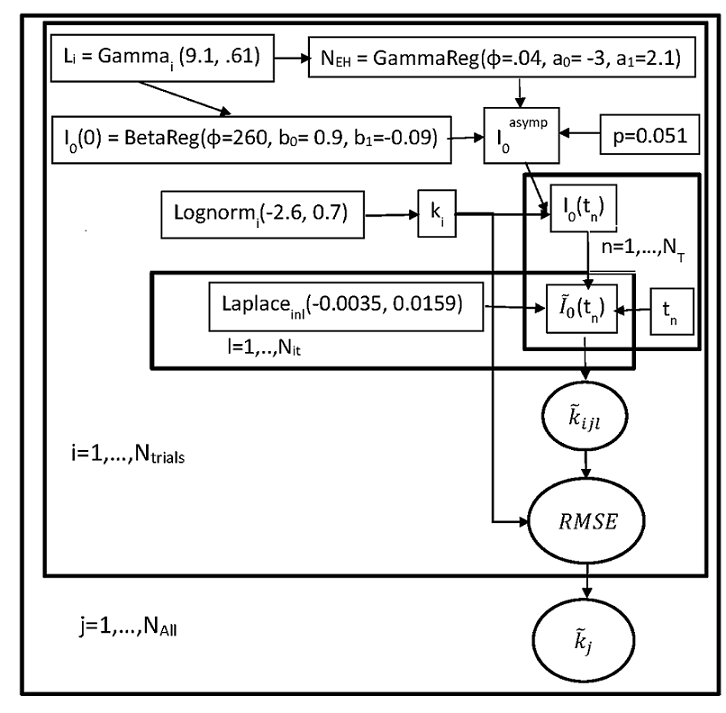
The tool provides a formula-based stochastic simulation strategy for TPS. We model the rate constant (lognormal),
measurement error (Laplace), peptide length (Gamma), relative abundance (RA) of the monoisotopic peak
(beta regression), and the number of exchangeable hydrogens (Gamma regression). RA and the number of exchangeable
hydrogens are correlated via the peptide length. Generalized regression models are used to model these parameters.
The models are used in simulations of the rate constant to minimize the root-mean-squared error.
This algorithm simulates time point selection for sampling dynamic proteome as described in:
Sadygov V. ,
Zhang W. , and
Sadygov RG,
Journal of Proteome Research; 2020, v. 19, 2105−2112.
To
download the code, please, provide your name, email and institution information.
Name :
(First*)
(Last*)
Email ID*:
Institution*:
* Please, provide the requested information.